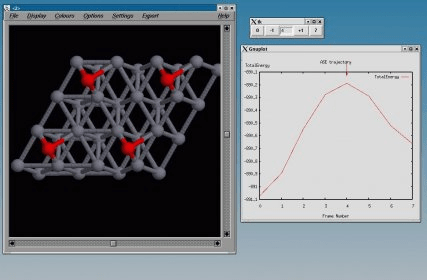
Dacapo is a total energy program based on density functional theory. It uses a plane wave basis for the valence electronic states and describes the core-electron interactions with Vanderbilt ultrasoft pseudo-potentials.
For an overview see the Pseudopotential_Library page. Calculations using dacapo are done using the Atomic Simulation Environment (ASE).

It is designed for visual and auditory event-related potentials acquisition.
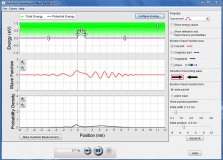
Explore the properties of the wave functions that describe these particles.

A simple free tool to convert between nearly all known units of density.





Comments